
CFCE is actively developing new analytical methods and pipelines to perform systematic analysis of epigenetic and transcriptional datasets. We currently use our in-house "CHIPS", "VIPER" and "CoBRA" pipelines for the initial mapping, peak calling, unsupervised and supervised analysis of epigenetic and RNA-seq datasets.
CHIPS pipeline
We developed a Snakemake pipeline called CHIPS (CHromatin enrIchment ProcesSor) to streamline the processing of ChIP-seq, ATAC-seq, and DNase-seq data. The pipeline supports single- and paired-end data and is flexible to start with FASTQ or BAM files. It includes basic steps such as read trimming, mapping, and peak calling. In addition, it calculates quality control metrics such as contamination profiles, polymerase chain reaction bottleneck coefficient, the fraction of reads in peaks, percentage of peaks overlapping with the union of public DNaseI hypersensitivity sites, and conservation profile of the peaks. For downstream analysis, it carries out peak annotations, motif finding, and regulatory potential calculation for all genes. The pipeline ensures that the processing is robust and reproducible. github.com/liulab-dfci/CHIPS
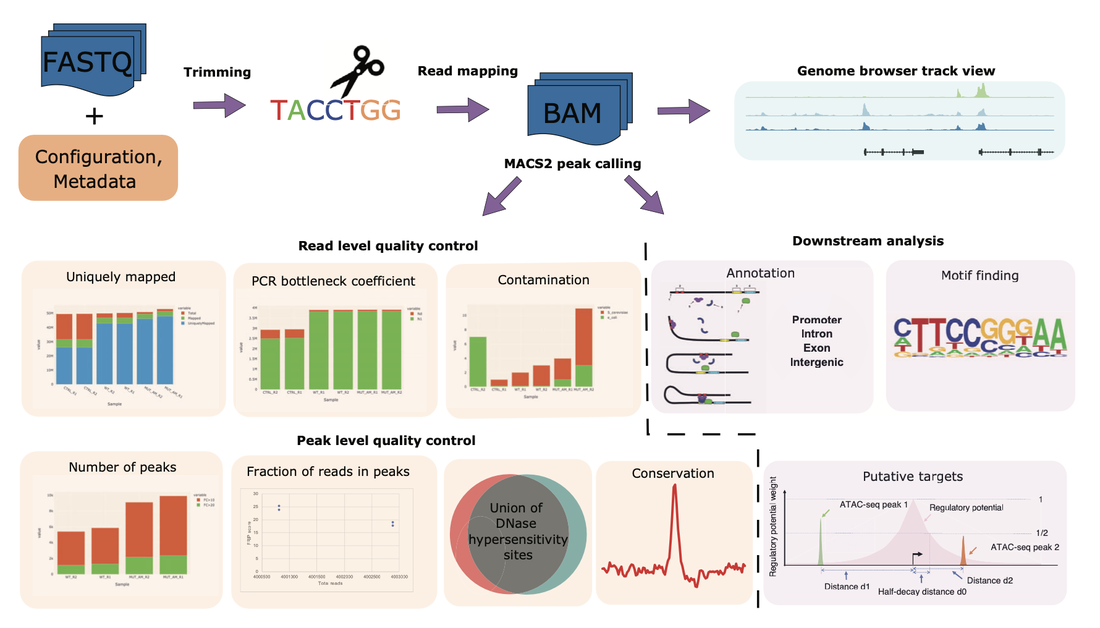 CHIPs pipeline
CHIPs pipeline
Reference:
Len Taing, Gali Bai, Clara Cousins, Paloma Cejas, Xintao Qiu, Zachary T. Herbert, Myles Brown, Clifford A. Meyer, X. Shirley Liu, Henry W. Long, Ming Tang. CHIPS: A Snakemake pipeline for quality control and reproducible processing of chromatin profiling data. F1000Research 2021, 10:517
Qian Qin, Shenglin Mei, Qiu Wu, Hanfei Sun, Lewyn Li, Len Taing, Sujun Chen, Fugen Li, Tao Liu, Chongzhi Zang, Han Xu, Yiwen Chen, Clifford A. Meyer, Yong Zhang, Myles Brown, Henry W. Long, X. Shirley Liu. ChiLin: a comprehensive ChIP-seq and DNase-seq quality control and analysis pipeline. BMC Bioinformatics 17, 404 (2016)
Len Taing, Gali Bai, Clara Cousins, Paloma Cejas, Xintao Qiu, Zachary T. Herbert, Myles Brown, Clifford A. Meyer, X. Shirley Liu, Henry W. Long, Ming Tang. CHIPS: A Snakemake pipeline for quality control and reproducible processing of chromatin profiling data. F1000Research 2021, 10:517
Qian Qin, Shenglin Mei, Qiu Wu, Hanfei Sun, Lewyn Li, Len Taing, Sujun Chen, Fugen Li, Tao Liu, Chongzhi Zang, Han Xu, Yiwen Chen, Clifford A. Meyer, Yong Zhang, Myles Brown, Henry W. Long, X. Shirley Liu. ChiLin: a comprehensive ChIP-seq and DNase-seq quality control and analysis pipeline. BMC Bioinformatics 17, 404 (2016)
VIPER pipeline
VIPER (Visualization Pipeline for RNA-seq analysis) is an analysis workflow that combines some of the most popular tools to take RNA-seq analysis from raw sequencing data, through alignment and quality control, into downstream differential expression and pathway analysis. VIPER has been created in a modular fashion to allow for the rapid incorporation of new tools to expand the capabilities. This capacity has already been exploited to include very recently developed tools that explore immune infiltrate and T-cell CDR (Complementarity-Determining Regions) reconstruction abilities. The pipeline has been conveniently packaged such that minimal computational skills are required to download and install the dozens of software packages that VIPER uses. bitbucket.org/cfce/viper/src/master/
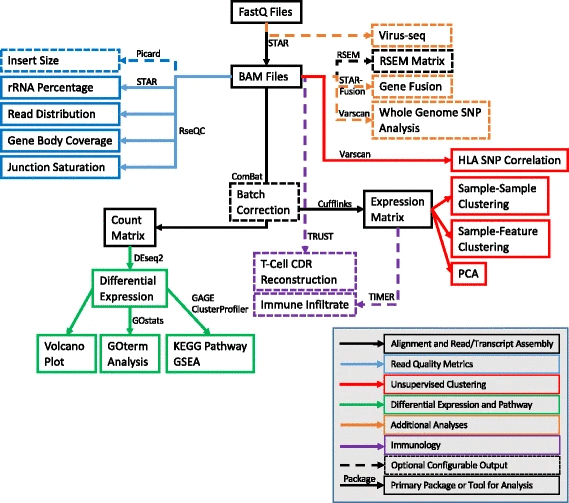
Reference:
Cornwell M, Vangala M, Taing L, Herbert Z, Köster J, Li B, Sun H, Li T, Zhang J, Qiu X, Pun M, Jeselsohn R, Brown M, Liu XS & Long HW. VIPER: Visualization Pipeline for RNA-seq, a Snakemake workflow for efficient and complete RNA-seq analysis. BMC Bioinformatics volume 19, 135 (2018)
Cornwell M, Vangala M, Taing L, Herbert Z, Köster J, Li B, Sun H, Li T, Zhang J, Qiu X, Pun M, Jeselsohn R, Brown M, Liu XS & Long HW. VIPER: Visualization Pipeline for RNA-seq, a Snakemake workflow for efficient and complete RNA-seq analysis. BMC Bioinformatics volume 19, 135 (2018)
CoBRA pipeline
CoBRA(Containerized Bioinformatics workflow for Reproducible ChIP/ATAC-seq Analysis) provides a comprehensive state-of-the-art ChIP-seq and ATAC-seq analysis pipeline that can be used by scientists with limited computational experience. This enables researchers to gain rapid insight into protein–DNA interactions and chromatin accessibility through sample clustering, differential peak calling, motif enrichment, comparison of sites to a reference database, and pathway analysis. CoBRA is publicly available online at https://bitbucket.org/cfce/cobra; cfce-cobra.readthedocs.io/en/latest/
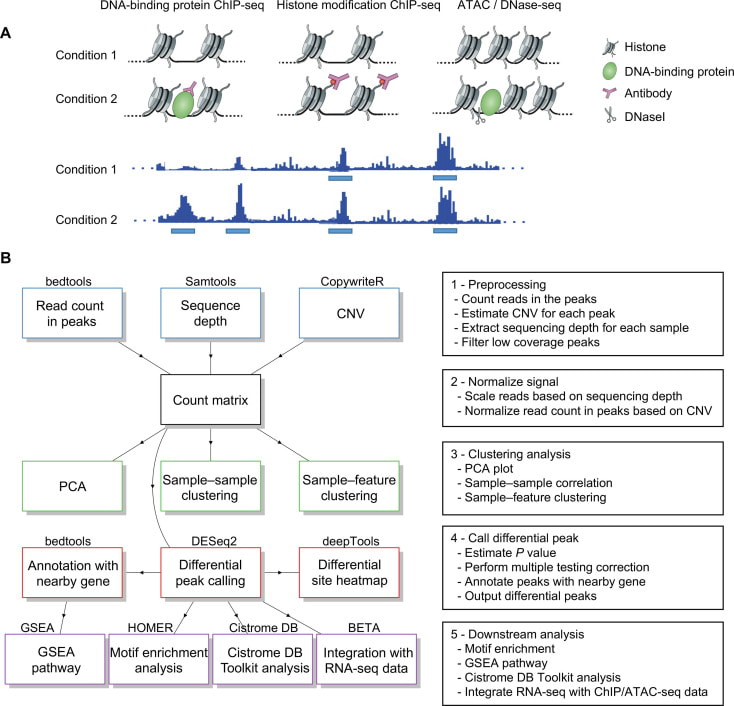
Reference:
Xintao Qiu, Avery S.Feit, Ariel Feiglin, Yingtian Xie, Nikolas Kesten, LenTaing, Joseph Perkins, Shengqing Gu, Yihao Li, Paloma Cejas, Ningxuan Zhou, Rinath Jeselsohn, Myles Brown, X.Shirley Liu, Henry W.Long. CoBRA: Containerized Bioinformatics Workflow for Reproducible ChIP/ATAC-seq Analysis. Genomics Proteomics Bioinformatics. 2021 Jul 17;S1672-0229(21)00154-6
Additional References applying CoBRA:
Xintao Qiu, Nadia Boufaied, ..., Henry W. Long & David P. Labbé. MYC drives aggressive prostate cancer by disrupting transcriptional pause release at androgen receptor targets. Nat Commun 13, 2559 (2022)
Xintao Qiu, Lisha G. Brown, ..., Henry W. Long and Eva Corey. Response to supraphysiological testosterone is predicted by a distinct androgen receptor cistrome. JCI Insight (2022)
Mark Pomerantz, Xintao Qiu, …Matthew Freedman. Prostate cancer reactivates developmental epigenomic programs during metastatic progression. Nat Genet 52, 790–799 (2020)
Yihao Li, Xintao Qiu, … Myles Brown. FGFR-inhibitor-mediated dismissal of SWI/SNF complexes from YAP-dependent enhancers induces adaptive therapeutic resistance. Nat Cell Biol 23, 1187–1198 (2021)
Xintao Qiu, Avery S.Feit, Ariel Feiglin, Yingtian Xie, Nikolas Kesten, LenTaing, Joseph Perkins, Shengqing Gu, Yihao Li, Paloma Cejas, Ningxuan Zhou, Rinath Jeselsohn, Myles Brown, X.Shirley Liu, Henry W.Long. CoBRA: Containerized Bioinformatics Workflow for Reproducible ChIP/ATAC-seq Analysis. Genomics Proteomics Bioinformatics. 2021 Jul 17;S1672-0229(21)00154-6
Additional References applying CoBRA:
Xintao Qiu, Nadia Boufaied, ..., Henry W. Long & David P. Labbé. MYC drives aggressive prostate cancer by disrupting transcriptional pause release at androgen receptor targets. Nat Commun 13, 2559 (2022)
Xintao Qiu, Lisha G. Brown, ..., Henry W. Long and Eva Corey. Response to supraphysiological testosterone is predicted by a distinct androgen receptor cistrome. JCI Insight (2022)
Mark Pomerantz, Xintao Qiu, …Matthew Freedman. Prostate cancer reactivates developmental epigenomic programs during metastatic progression. Nat Genet 52, 790–799 (2020)
Yihao Li, Xintao Qiu, … Myles Brown. FGFR-inhibitor-mediated dismissal of SWI/SNF complexes from YAP-dependent enhancers induces adaptive therapeutic resistance. Nat Cell Biol 23, 1187–1198 (2021)
scATAnno
scATAnno is a workflow designed to automatically annotate scATAC-seq data using large-scale scATAC-seq reference atlases. This workflow can generate scATAC-seq reference atlases from publicly available datasets, and enable accurate cell type annotation by integrating query data with reference atlases, without the aid of scRNA-seq profiling. https://scatanno-main.readthedocs.io/
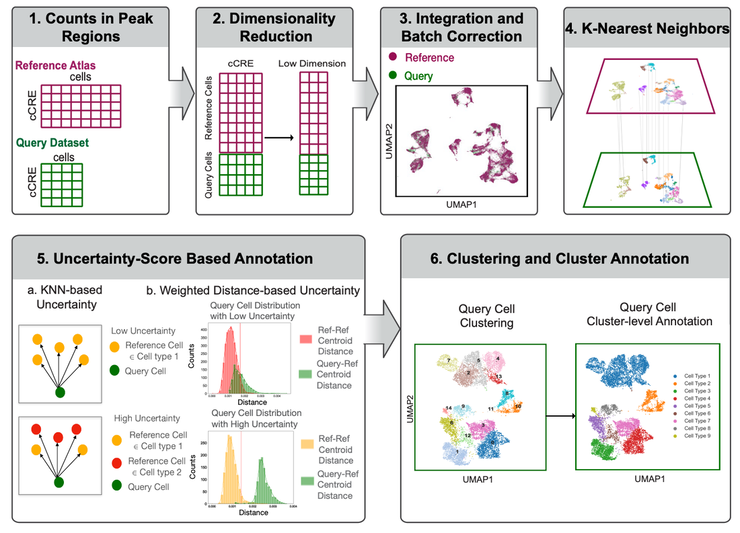
Reference:
Yijia Jiang, Zhirui Hu, Junchen Jiang, Alexander Zhu, Yi Zhang, Allen Lynch, Yingtian Xie, Rong Li, Ningxuan Zhou, Cliff A. Meyer, Paloma Cejas, Myles Brown, Henry W. Long, Xintao Qiu. scATAnno: Automated Cell Type Annotation for single-cell ATAC sequencing Data. bioRxiv 2023.06.01.543296.
Yijia Jiang, Zhirui Hu, Junchen Jiang, Alexander Zhu, Yi Zhang, Allen Lynch, Yingtian Xie, Rong Li, Ningxuan Zhou, Cliff A. Meyer, Paloma Cejas, Myles Brown, Henry W. Long, Xintao Qiu. scATAnno: Automated Cell Type Annotation for single-cell ATAC sequencing Data. bioRxiv 2023.06.01.543296.